

Most market-entry delays we see come from evidence gaps, not missing security work: unclear system boundaries, weak traceability, and documents that can’t be tied to a specific product release.
This article shows you the minimum viable cybersecurity evidence to prepare for EU MDR and NHS DTAC expectations and how to build it once, so it can be reused across routes to market.
A Mantra Systems x Cyber Alchemy Perspective - Episode 1
This article is published in partnership with Cyber Alchemy. Cyber Alchemy focuses on cybersecurity, helping teams develop and evidence security for software-enabled and connected medical devices. Mantra Systems specialises in regulatory strategy and clinical evidence for UKCA and EU MDR/IVDR pathways. Together, we're producing a practical series for MedTech teams: what to build, what to defer, and how to avoid avoidable rework when moving between UK, NHS procurement, and EU routes.
Key takeaways
- EU MDR cybersecurity requirements are more explicit than the current GB framework. For EU market entry, manufacturers need to explain the IT and security conditions required for safe use and demonstrate that cybersecurity has been built into software development, testing, and risk management. In the UK, cybersecurity expectations will show up through procurement, especially NHS routes using DTAC, even if your UKCA pathway feels less prescriptive than EU MDR wording.
- Building a single versioned Security Evidence Pack can reduce duplication of costs and effort across EU MDR, UKCA, and DTAC. The core work is done once, then tailored for each route, which means fewer document rewrites, fewer repeated testing exercises, and fewer delays caused by missing or inconsistent evidence.
- Your evidence must be release‑tied: test reports, SBOM, vulnerability handling and residual risks should clearly reference what you are placing on the market now.
- If you want a fast way to benchmark your gaps, use the two free downloads near the end of this article.
Companion perspective (Mantra Systems)
This article focuses on cybersecurity evidence (what to build, how to evidence it, and how to keep it release-tied). The companion article from Mantra Systems covers the clinical and regulatory side of the same decision: classification, pathway choice (UK vs EU), and Minimum Viable Evidence beyond cybersecurity. Read both to align your cyber evidence plan with the right regulatory route and claim set.
Ready for the next step? Book a joint Cyber Alchemy × Mantra Systems review to map your route and evidence plan.
Why this matters now
1. “UK first” is not one route; it’s two (private practice vs NHS)
Many founders we work with opt to launch in the UK first, as their home market. Frequently, there is an assumption that means “UKCA plus a few security questionnaires during procurement”.
In reality, UK market entry often splits into two very different cyber assurance paths:
- Private practice/private providers: requirements are usually contractual and variable (supplier due diligence questionnaires, requested cybersecurity penetration test summaries, security policies, incident response posture, etc.). There’s no single national “one form to rule them all”.
- NHS/NHS-adjacent procurement: DTAC is the standardised entry point for procurement and due diligence (including pilots), with evidence expected to be supplied by the manufacturer and kept up to date as the product changes.
This matters because DTAC cybersecurity requirements often push you to demonstrate organisational cyber posture (as well as product‑level controls) earlier than you expect, and that can delay pilots if left until late.
2. EU MDR and DTAC
It’s easy to assume DTAC and EU MDR are “equivalents” because both lead to long security conversations — but they’re not the same gate.
- DTAC (NHS procurement assurance) is a buying and due diligence requirement. It helps NHS organisations assess whether a digital product is safe to adopt and operate. DTAC tends to focus on both organisational posture (your ability to operate securely) and product controls (testing, patching, monitoring, incident response), often early in pilots and procurement.
- EU MDR (CE marking) is a regulatory requirement for market access. It focuses on safety and performance for the intended purpose, as well as the technical documentation required to place the device on the EU market (with cybersecurity expectations embedded in Annex I/GSPR).
The good news is that the evidence overlaps. If you build a robust security system and evidence it well, you can “wrap” it for DTAC or EU MDR without rebuilding the underlying truth.
3. DTAC has a hard transition date in 2026
NHS England has issued an updated DTAC form (version 2.0 updated 24 February 2026). The previous DTAC form should not be used from 6 April 2026 onwards.
If NHS is in your near-term plan, treat DTAC readiness as a programme with artefacts, owners, and a refresh cadence, not a last-minute questionnaire exercise.
Further reading: DTAC support from Cyber Alchemy.
4. GB still recognises CE for now — and may recognise it indefinitely
The Medicines and Healthcare products Regulatory Agency (MHRA) regulates medicines and medical devices in the UK and has published updated timelines showing CE acceptance in Great Britain running (in many cases) to 30 June 2030, and is currently consulting on indefinite recognition of CE-marked devices in GB (consultation closes 10 April 2026).
Translation: your first-market decision should consider where your cybersecurity investment, and crucially the evidence of that investment, gives you the most optionality.
Note on terminology: In this article, Great Britain (GB) means England, Scotland and Wales. That matters because medical device regulation differs across the UK following Brexit. Northern Ireland follows EU rules for placing devices on the NI market, while GB uses the UK framework (UKCA routes). When we say “GB recognises CE for now”, we’re referring specifically to GB market access arrangements.
UKCA vs EU MDR: why cybersecurity “feels” different
Founders often hear “EU is stricter on cybersecurity” and interpret that as “UK market access will be easy”. That’s not what’s happening. The differences are mostly about how explicit the requirements are in legislation and how evidence is assessed in practice.
EU MDR cybersecurity requirements: what GSPR 17.2 and 17.4 mean in practice
The EU MDR includes an explicit requirement that manufacturers set out the minimum requirements for hardware, IT network characteristics, and IT security measures, including protection against unauthorised access, needed to run the software as intended (GSPR 17.4). It also calls for a “state of the art” approach to software and electronic programmable systems, including the development life cycle, risk management (including information security), and verification and validation (GSPR 17.2).
Explainer: In regulatory terms, “state of the art” does not mean cutting-edge technology for its own sake. It means using current, generally accepted industry practice for your device type and risk profile — typically demonstrated through recognised standards, established engineering controls, and documented justification for any deviations.
Rather than creating a “burden”, these clauses provide a clear framework for what “good” looks like: you are expected to define the environment your product is designed for, design security in a way that is proportionate to the risks, and be able to demonstrate (with evidence) that the controls work for the release you are placing on the market. In practice, that means documenting your intended environment assumptions (identity model, network conditions, supported platforms, update mechanism), setting out the minimum security expectations needed for safe operation, and maintaining a traceable line from threats → controls → verification evidence that a reviewer can follow quickly.
In practice, EU MDR reviewers tend to look for the following evidence signals:
| EU MDR requirement signal | What you show (evidence examples) |
|---|---|
| Minimum IT/security requirements (intended environment) | A short ‘minimum IT requirements’ section: identity model, network assumptions, supported platforms, update mechanism, logging expectations, secure configuration guidance. |
| State of the art lifecycle + V&V | Secure development lifecycle controls, verification evidence index and release gating. |
| Risk management, including information security | Threat model and security risk register, aligned to safety and performance risk management where relevant. |
| Verification of adopted solutions | Penetration test and vulnerability test summaries scoped to boundary, remediation actions and re-test confirmation. |
| Post-market maintainability | Vulnerability monitoring, SBOM coverage statement, security PMS cadence and triggers for re-test/impact assessment. |
A key nuance: guidance tells you what to do (“implement access control”, “manage vulnerabilities”, “protect against unauthorised access”). The time and cost usually surface when teams have to work out how to implement those requirements in a way that is proportionate to their threat model, realistic for clinical deployment, and clearly evidenced to a reviewer. That “how” is where submissions and procurements often slow down and where getting it right early avoids expensive rework later.
UKCA (GB) is currently less prescriptive in the wording — but still evidence-driven in practice
Under the current UK MDR 2002 framework, you still need to demonstrate conformity with the applicable requirements through the correct conformity assessment route, and (depending on class) via a UK Approved Body.
What’s different is that the current GB pre-market framework does not (yet) present cybersecurity as a standalone “controls checklist” clause, as people experience in the EU MDR’s explicit GSPR language. In practice, cybersecurity is assessed through existing safety/performance and software validation expectations (“state of the art” lifecycle, risk management, verification and validation), as well as what you communicate to users about safe installation, interoperability, and residual risks.
The UK has signalled it intends to move closer to EU-style explicit SaMD cyber requirements
The UK’s SaMD consultation outcome states that the current regulations contain few provisions specifically aimed at SaMD/AIaMD, and that the government intends to introduce a requirement akin to EU MDR GSPR 17.4, including minimum requirements for security measures and protection against unauthorised access.
Practical takeaway: even if you start with a GB route, building your evidence pack so it maps cleanly to EU MDR expectations reduces future-market rework.
The business reality: cybersecurity affects time-to-market (especially in the EU)
A quick clarification: regulation vs procurement are different “gates”.
It’s helpful to separate two things that often get mixed up:
- Regulatory market access (e.g., EU MDR for CE marking, and the UK MDR 2002 / UKCA framework for GB) determines whether you can legally place a device on a market.
- Procurement assurance (e.g., NHS DTAC) determines whether an organisation is willing and able to adopt your product in practice.
They overlap heavily in the evidence they rely on, but they’re not the same process. This is why we recommend building one core Security Evidence Pack that can be packaged both for regulatory documentation and for procurement due diligence.
Device classification applies equally to cybersecurity: regulatory class and pathway decisions affect the volume and rigour of documentation, fees and time to market. If you need a refresher, Mantra Systems’ article on EU vs US classification explains why early classification errors create expensive rework.
For EU entry specifically, notified body capacity is still a pacing factor. The European Commission’s 14th notified body survey (data up to 28 February 2025) shows many notified bodies reporting 13–18 months as the most common band to reach a new MDR certificate (QMS and QMS+product), and it highlights that a substantial share of time is often with the manufacturer (document revision/readiness).
That’s exactly why this guide focuses on Minimum Viable Cyber Evidence: to reduce avoidable back-and-forth.
What good looks like
Good market-ready cyber evidence has a single source of truth: one Security Evidence Pack that is:
- Versioned to a specific product release
- Built on stable system boundaries (device/app/cloud/integrations)
- Traceable (threats → controls → verification → evidence location)
- Maintainable (clear refresh triggers so it doesn’t rot)
Then you present the same truth differently depending on the audience:
- GB/UKCA packaging: demonstrate conformity via technical documentation and the appropriate conformity route (including UK Approved Body involvement where required).
- EU MDR packaging: map explicitly to Annex I/GSPR expectations (including minimum IT/security requirements) and to MDCG cybersecurity guidance.
- NHS procurement: DTAC-aligned summaries plus supporting evidence (and often DSPT alignment where applicable).
You’re not changing the truth, you’re changing navigation.
Minimum Viable Cyber Evidence checklist
This is the smallest set that reliably supports private UK due diligence → NHS DTAC → EU packaging, without overbuilding.
1. Security architecture and boundaries
- Boundary diagram: device/app/cloud/APIs/integrations + trust boundaries
- Data flows + where data is stored/processed
- Responsibilities: what you control vs what the customer controls
2. Threat model and risk register
- Stated assumptions
- Top abuse cases
- Threat modelling should be aligned with a recognised framework, such as STRIDE.
3. Risk-control traceability matrix
A single table linking:
- Threat/risk → mitigation → design requirement → verification evidence
- This table shows that you have considered threats, designed and implemented mitigations into the product, and then verified, through practical means, that they have addressed the risk as intended.
4. Verification evidence index
A controlled index of evidence artefacts (reports, test outputs, configs, procedures), each tied to:
- Product release/version
- Owner
- Location
5. Security testing evidence
- What was tested, when, and why the scope matches the boundary
- Findings summary
- Remediation actions and re-test confirmation to confirm that all unacceptable risks have been addressed
6. SBOM and vulnerability management
- SBOM coverage statement (what is included/excluded)
- CVE monitoring workflow and triage log
- One worked example from detection → decision → fix/mitigation → closure
7. Secure update and patch policy
- How updates are delivered (signing, rollback/failsafe as applicable)
- Routine vs urgent timelines you can actually meet in clinical deployment
8. Post-market cyber assurance cadence
- What triggers a security impact assessment
- When you re-test
- How evidence stays aligned with shipped versions
9. NHS layer (only if NHS is a target market)
DTAC technical assurance commonly expects cybersecurity evidence around:
- Cyber Essentials position
- Penetration/vulnerability testing evidence
- Clarity on internal vs external testing
- Adherence with the DSIT Software Security Code of Practice that governs secure design and development, secure build environment, secure deployment, maintenance and communication with customers
- Defined logging and reporting requirements
This is the checklist we use to keep procurement and assurance work from becoming a last-minute scramble. It covers the ten artefacts that are most frequently requested — including security architecture and boundaries, threat model and risk register, a risk-control traceability matrix, SBOM and vulnerability monitoring, a VDP/PSIRT route, secure update/patch policy, security test evidence, logging/monitoring approach, supplier security controls, and an incident response playbook with rehearsal evidence — plus a suggested cadence for keeping them current.
Free download: 10 Core Procurement Artefacts
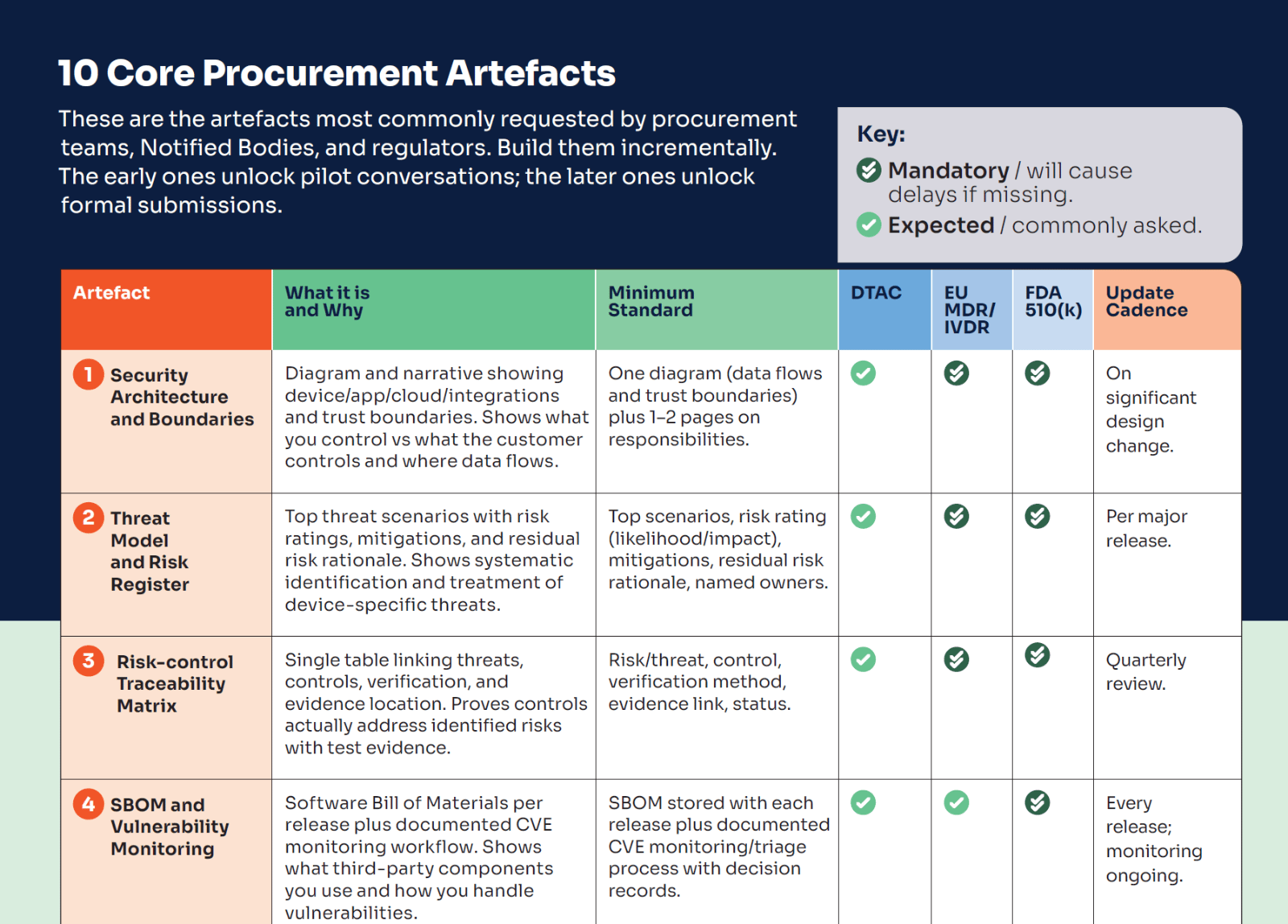
Five common failure patterns that cause delays (and how to avoid them)
These aren’t theoretical. They are repeatable patterns that slow procurement and submissions because they break the “evidence story”.
| Failure pattern | What to do instead |
|---|---|
| Unclear system boundaries (scope ambiguity poisons threat modelling, test scope and evidence packaging) | Create system architecture documentation and data flow diagrams. Assign trust boundaries and a responsibility matrix, then scope all evidence to it. |
| Traceability gap (you can’t show threat → control → verification cleanly) | Maintain a short, traceable table listing only the top threats, tied to the release you are shipping. STRIDE is a good framework to use for threat modelling. |
| SBOM exists once but isn’t release‑tied | Automate SBOM generation per release and log ‘are we affected?’ decisions for material vulnerabilities. |
| Patch policy commitments you can’t keep | Define realistic timelines that align with clinical deployment and explain how you manage exceptions. |
| External developers weren’t contracted to produce evidence | Make security requirements and evidence deliverables part of the definition of done and the contract. |
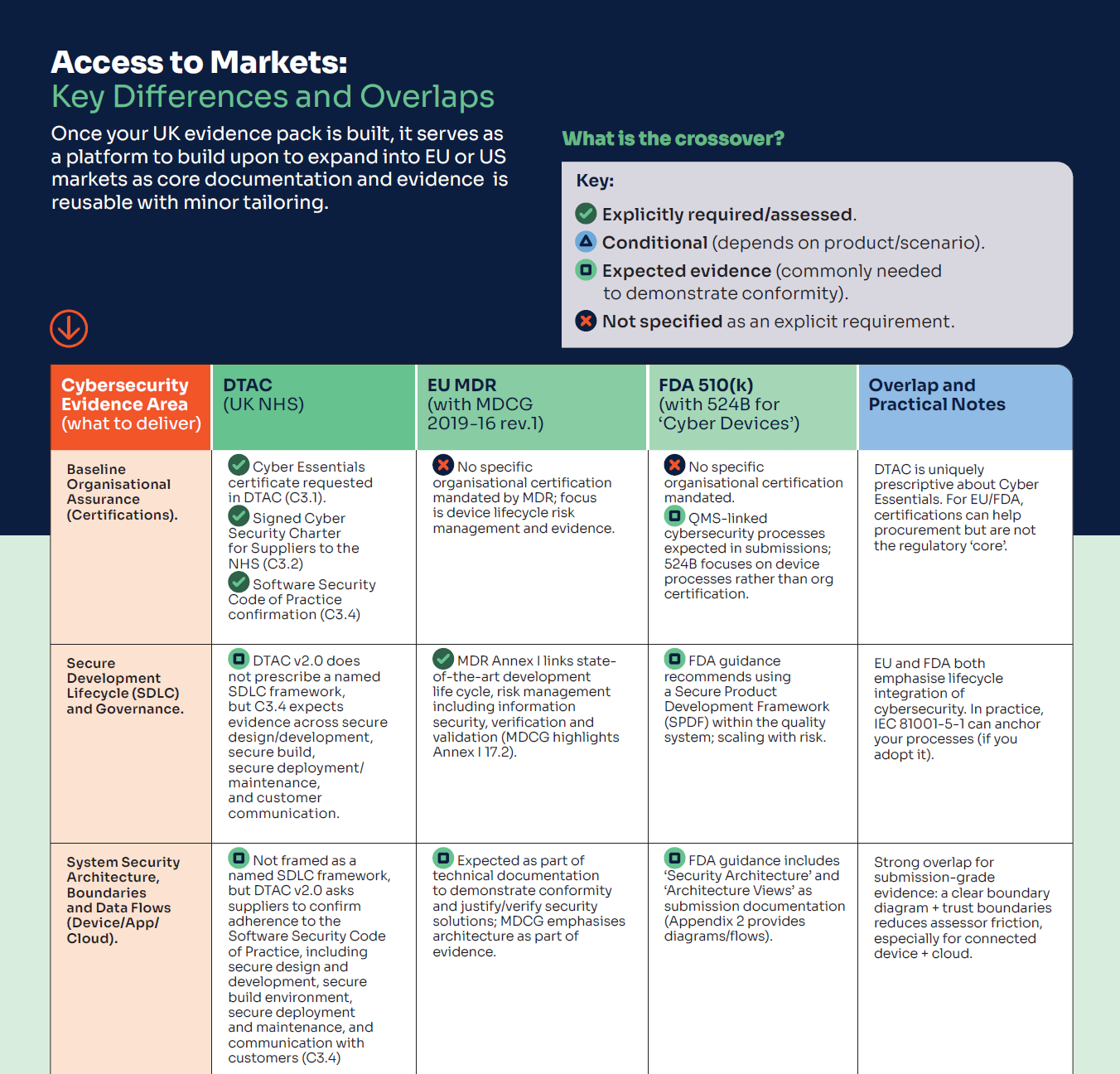
If you want a quick reference you can actually use while planning your roadmap, we’ve put together a tri-fold PDF that summarises the key cybersecurity evidence differences and overlaps across the UK, EU and US — including the practical split in the UK between private practice and NHS procurement (DTAC). It’s designed to help you answer two questions fast: what evidence can I build once and reuse, and what needs tailoring for the market I’m targeting first (with the cost/time implications in mind).
Free download: Market Differences: UK vs EU vs US
Mantra Systems’ companion article provides the founder decision framework for the UK vs EU, as well as the broader clinical/regulatory evidence pathways, so you can plan your route to market with the right clinical evidence and pathway.
Next step: Book a joint review
If you want a clear, evidence-first view of what will satisfy assessors and procurement teams (and what will be wasted effort), book a review with Cyber Alchemy and Mantra Systems.
In 30 minutes we’ll:
- Discuss your route to market, drawing on our experts’ real-world experience
- Identify any key challenges you might face on your route to market and how to overcome them
- Advise on what to build now vs what to defer (avoid “future-market overbuild”)
FAQs (EU MDR and DTAC cybersecurity requirements)
What are the EU MDR cybersecurity requirements for software medical devices?
EU MDR expects you to define minimum IT/security requirements for the intended environment (GSPR 17.4) and demonstrate a state‑of‑the‑art approach to lifecycle, risk management (including information security), verification and validation (GSPR 17.2). In practice, reviewers look for a traceable evidence story tied to the release you are putting on the market.
What are DTAC cybersecurity requirements and who needs to meet them?
DTAC is used in NHS procurement and due diligence. If you intend to sell to, pilot with, or integrate with NHS organisations, you should expect to provide DTAC‑aligned evidence covering organisational posture and product‑level security controls.
How do DTAC requirements differ from private practice procurement checks?
Private providers typically use variable supplier questionnaires. DTAC is more standardised and tends to require more repeatable, auditable evidence that you can keep current as your product changes.
Does UKCA marking require an SBOM?
SBOM is not typically presented as a named, standalone UKCA pre‑market requirement in the way it is in some US contexts, but it is a strong evidence signal for supply chain control and post‑market vulnerability handling, and is increasingly expected in procurement.
How often should penetration testing be repeated?
There is no single universal interval, though at least annual intervals are generally considered a reasonable approach. What really matters is a defensible cadence plus triggers tied to change (major releases, new integrations, significant architectural changes, or new vulnerability classes).
How do we keep evidence from going stale after go‑live?
Treat your Security Evidence Pack as a maintained asset: assign owners, define refresh triggers, and tie updates to release management and post‑market monitoring.
About the author
Luke Hill, Senior Security Consultant at Cyber Alchemy
Luke brings deep expertise in security consultancy, penetration testing and regulatory‑aligned security measures in the Health and Social Care sector. He leads Cyber Alchemy’s technical and regulatory efforts in the MedTech space, supporting a broad range of MedTech companies in building resilient devices and applications that comply with complex UK, US, and EU regulations.
Related
Case study: See how Cyber Alchemy supported Adaptix with MedTech cybersecurity and assurance work in practice
- Episode 1 from Mantra Systems
- Where to Launch First? A MedTech Founder's Regulatory Roadmap to the EU, UK and US
- Episode 1 from Cyber Alchemy
- EU MDR & NHS DTAC Cybersecurity Requirements for UK Market Entry
- Episode 2 from Mantra Systems
- How to handle non-conformities and get back on track
- Episode 2 from Cyber Alchemy
- EU MDR, FDA 510(k) and DTAC Cybersecurity Nonconformities: How to Recover















































































































{kind=link}