

Medical device vigilance is a two-way street. It safeguards patients from harm and protects manufacturers from reputational damage and financial losses.
Vigilance is crucial because even the most meticulously designed medical device can encounter unforeseen challenges in the real world. Vigilance acts as a critical feedback loop, providing invaluable insights that drive product improvement, strengthen regulatory compliance, and protect patients from harm.
The Critical Role of Vigilance in Medical Devices
But vigilance is not just good practice – it is a regulatory requirement in most major markets. Regulatory bodies like the FDA in the US, the European Commission in the EU, and the MHRA in the United Kingdom mandate that medical device manufacturers have robust vigilance systems in place. These systems must include procedures for collecting, analysing, and reporting incidents to the relevant authorities.
Failing to comply with these requirements can lead to serious consequences, including product recalls, fines, and even legal action. For example, in 2019, a notable case involving a medical device manufacturer resulted in a $15 million fine due to failure to report a product recall to the FDA, illustrating the severe repercussions of neglecting post-market vigilance obligations.
What Vigilance Entails: A Closer Look
Vigilance is the ongoing, systematic process of monitoring the safety and performance of medical devices throughout their entire lifecycle. It includes diligently collecting and analysing data on any incidents or adverse events that may occur. Think of it as a safety net, constantly catching and evaluating potential risks to ensure that medical devices remain effective and, above all, safe for the patients who rely on them.
Incident reporting, a crucial component of vigilance, involves the detailed documentation and communication of any events that may impact the safety or performance of a medical device.
Accurate and timely incident reporting is essential for effective vigilance. It provides the data needed to identify trends, understand root causes, and take corrective actions to prevent similar incidents from occurring in future.
How to Build a Robust Vigilance System: A Practical Guide
Creating a robust vigilance system requires a structured approach and a commitment to continuous improvement. Here is a practical guide to get you started:
- Identify Relevant Regulations and Standards: The regulatory landscape for medical devices is constantly changing, with new requirements and updates emerging frequently. The introduction of the European Union Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR), for instance, has brought about significant changes in post-market surveillance requirements. In Great Britain, the MHRA introduced its own post‑market surveillance framework, which came into force on 16 June 2025 and includes more prescriptive obligations than those previously in place. Similarly, the FDA’s ongoing efforts to modernise medical device regulations in the US add another layer of complexity. Companies must proactively monitor regulatory updates (such as those shared in our regulatory newsletter), participate in workshops and conferences, and engage with regulatory bodies to ensure ongoing compliance.
- Establish Internal Reporting Procedures: Develop clear and concise internal reporting procedures that outline the steps for reporting incidents, define roles and responsibilities, and establish communication channels. Ensure that all employees understand their role in the vigilance system.
- Develop a Comprehensive Incident Reporting Form: Create a user-friendly incident reporting form that captures all essential information. Include fields for patient demographics, device information, incident description, severity level, and any contributing factors. Make the form readily accessible and easy to use.
- Train Employees on Vigilance Procedures: Conduct regular training sessions to educate employees on vigilance procedures and the importance of incident reporting. Foster a culture of transparency and encourage proactive reporting.
- Implement a System for Data Management and Analysis: Invest in a system that can efficiently collect, store, and analyse vigilance data. This could be a dedicated software solution or a combination of tools and processes. Utilise data analytics to identify trends, predict potential issues, and generate insightful reports.
- Establish a Process for Corrective and Preventive Action (CAPA): Develop a systematic process for investigating incidents, identifying root causes, and implementing corrective and preventive actions. Ensure that CAPA activities are documented and tracked to completion.
- Regularly Review and Update Your Vigilance System: Vigilance is an ongoing process. Consistently review and update your system to ensure it remains effective and compliant with evolving regulations and industry best practices.
Where Vigilance Gets Complex: Navigating Global Requirements
Vigilance requirements can be complex, especially for companies operating in multiple markets. The US, EU, and Asian countries, for instance, have distinct regulations and reporting procedures. Understanding these nuances is crucial for ensuring compliance and avoiding costly penalties.
Here are some key differences to consider:
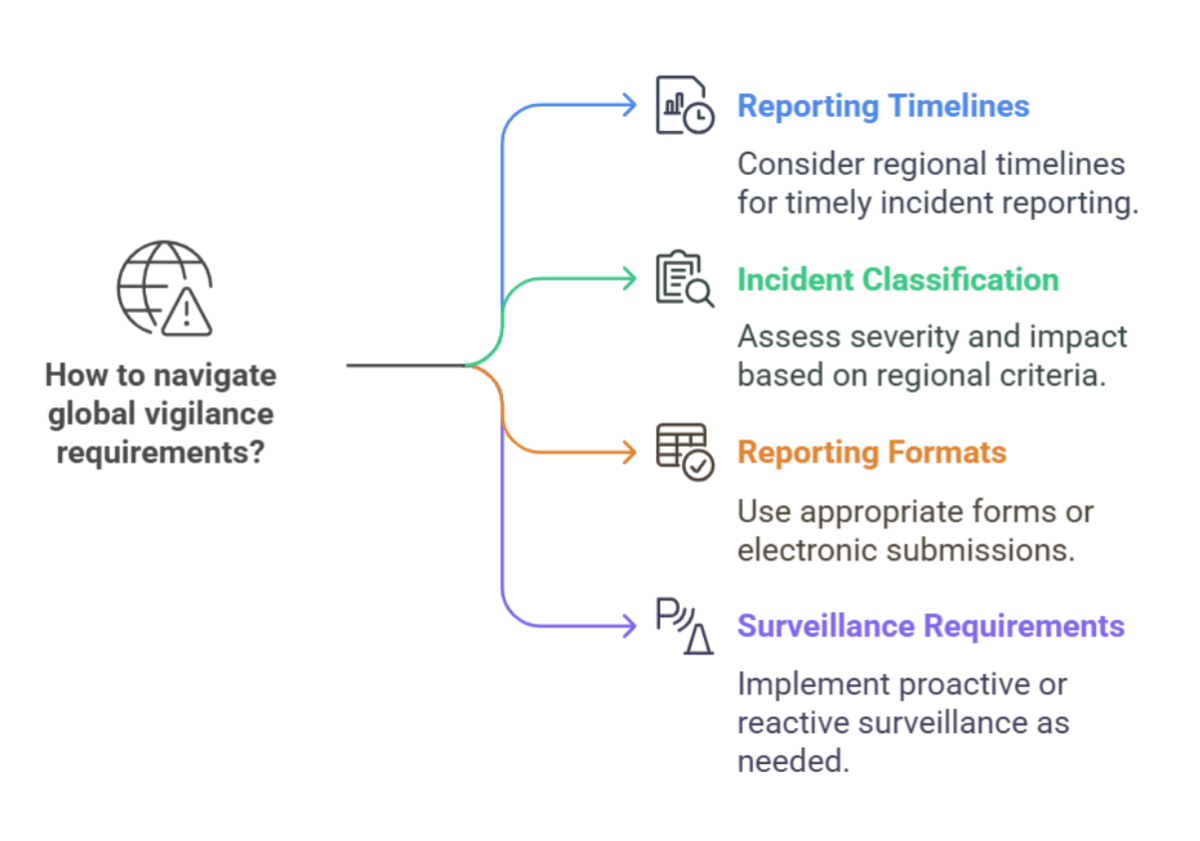
- Incident reporting timelines: Different regions may have different timelines for reporting incidents. For example, the EU MDR requires manufacturers to report serious incidents within 15 days, while the FDA has different timelines depending on the event’s severity.
- Incident classification: The criteria for classifying incidents may vary. Some regions may have more detailed classification systems, requiring manufacturers to assess the severity, impact, and probability of recurrence.
- Reporting formats: Different regions may have specific reporting forms or electronic submission requirements.
- Post-market surveillance requirements: The scope and depth of post-market surveillance activities may vary. Some regions may require more proactive data collection and analysis, while others rely more on reactive reporting.
Why Expert Guidance Matters
Building a robust vigilance system is complex, especially for companies with limited resources or those entering new markets. Mantra Systems, specialising in medical device market access, offers expert guidance to navigate vigilance and incident reporting. We assist with developing and implementing tailored vigilance systems, staying up-to-date with evolving regulations, managing complex data and reporting requirements, and ensuring compliant market entry into new regions. Leveraging Mantra Systems’ expertise streamlines vigilance efforts, reduces risks, and allows companies to focus on developing innovative medical technologies.
References:
European Union. Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices. Official Journal of the European Union. 2017;L117:1-175. Accessed August 11, 2025. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32017R0745
World Health Organization. Medical Device Vigilance: Guidance for Healthcare Professionals. 2017. Accessed August 11, 2025. https://www.who.int/tools/health-products-regulation-and-prequalification-learning-catalogue/vigilance
Global Harmonization Task Force, Study Group 2. Medical Devices Post Market Surveillance: Global Guidance for Adverse Event Reporting for Medical Devices. GHTF; 2006. Accessed August 11, 2025. https://www.imdrf.org/documents/ghtf-final-documents/ghtf-study-group-2-post-market-surveillancevigilance
European Commission. Guidance on the Vigilance System for Medical Devices. 2013. Accessed August 11, 2025. https://ec.europa.eu/docsroom/documents/32305/attachments/1/translations/en/renditions/native
Gupta P, Janodia MD, Jagadish PC, Udupa N. Medical device vigilance systems: India, US, UK, and Australia. Med Devices (Auckl). 2010;3:67-79. Accessed August 11, 2025. https://www.dovepress.com/article/download/5714












































































































