

In today’s ever-evolving Medical Device regulatory landscape, staying compliant with the European Union Medical Device Regulations (EU MDR) is crucial for businesses operating in or with the EU. However, navigating its complex and ever-changing framework can feel like a maze.
If you’re uncertain about the readiness of your EU MDR documentation, this article provides an overview of the essential steps to ensure you’re on track, from understanding key regulations to identifying potential gaps in your compliance process.
Grasping the Essentials: Key EU MDR Requirements You Need to Know
Compliance is accomplished by demonstrating conformity of your device with all relevant aspects of the EU MDR. Manufacturers are required demonstrate this compliance to gain regulatory approval for their medical devices.
Medical devices must be designed, manufactured, distributed, and tracked according to EU MDR requirements, and manufacturers must develop a suite of compliant regulatory systems, processes, and documents to continually monitor the safety and performance of their products.
This process begins with a detailed understanding of the regulation and the obligations it imposes upon manufacturers. A key part of demonstrating compliance is ensuring that your technical file includes everything required under the regulation. This consists of clinical evaluations, risk management documentation, and information on manufacturing processes. Being thorough at this stage ensures you don’t run into roadblocks later.
Conquering Your Technical Documentation: A Thorough Review is Essential
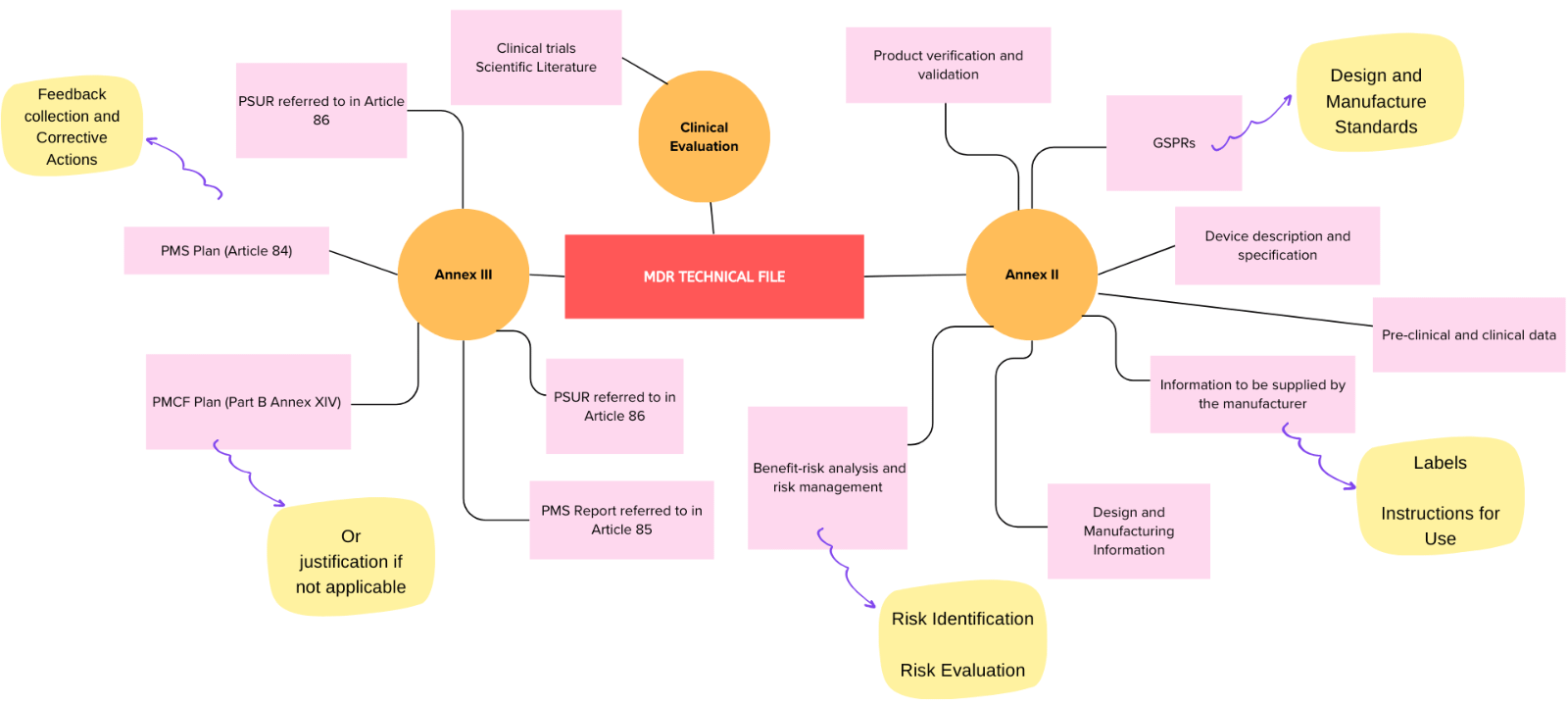
One of the most essential aspects of EU MDR compliance is your technical documentation. The regulation requires manufacturers to maintain detailed files that provide evidence of the safety, performance, and risk management of their products. These files must be available for review by Notified Bodies during audits and inspections.
Regulatory systems, processes, and documents required for compliance are more extensive than those necessary under the outgoing Medical Device Directive (MDD). Those required for MDR Compliance include the following:
- Quality Management System (QMS) covering the activities of the entire organisation
- Post-Market Surveillance (PMS) system monitoring safety and performance of each device following release onto the market
- Post-Market Clinical Follow-up (PMCF) and Vigilance system for each device
- Risk Management system to the standard in ISO 14971:2019 or equivalent
- Clinical Evaluation process for every medical device, documented as a State-of-the-Art Report (SOTA), Clinical Evaluation Report (CEP), and a Clinical Evaluation Report (CER) which must be continuously updated at appropriate intervals
Carefully reviewing each section of your technical file is essential. Missing even a small piece of documentation can cause delays or, worse, non-compliance. Successful compliance is demonstrated by the application of a CE-mark to the device.
Class I manufacturers (except for devices that are sterile or have a measuring function) may self-apply a CE-mark after producing a declaration of conformity. For all other classes of medical devices, a CE-mark may only be affixed once a Notified Body has issued a certificate of conformity following a regulatory review according to rules in Chapter IV of the MDR.
Navigating Special Cases: Ensuring Conformity with EU MDR Requirements
Certain devices, particularly those that include medicinal substances or components of animal or human origin, have additional requirements under the EU MDR. These special cases require detailed documentation to ensure compliance with stricter regulations. It’s important to identify if your device falls under these categories and ensure that you are meeting the additional regulatory requirements.
This can often be a challenging aspect of compliance. For example, if your device uses a medicinal substance or has biological components, it may be subject to additional testing and reporting obligations, e.g., a medicinal dossier. Understanding these specifics early on in the process can save you time and prevent future compliance hurdles.
Stay Compliant: The Importance of Regularly Updating Your Documentation
EU MDR compliance is an ongoing process. It’s not enough to simply submit your documentation and wait for approval. Once your device is on the market, continuous monitoring through PMS and PMCF is required. You must regularly update your technical file to reflect any changes, including modifications to the device, new clinical data, or updates to regulatory requirements.
Additionally, your clinical evaluations should be regularly reviewed to ensure that new evidence supports the ongoing safety and performance of your device.
Ensuring You’re Ready: How to Close the Gaps
With so many requirements and details involved, it can be easy to feel unsure about whether your documentation is truly ready for submission. Even the most diligent teams can miss critical elements or overlook specific areas of compliance.
This is where an automated tool can be a game-changer. An EU MDR compliance checker can help you quickly identify any gaps or discrepancies in your technical documentation. By conducting a comprehensive review of your files, you ensure that your submission is as complete and accurate as possible.
No Technical File, no CE mark, no access to the EU market.
Mantra Systems’ EU MDR Compliance Checker is built to take the guesswork out of the process. It flags any discrepancies and offers a report card for compliance gaps, providing you with reassurance that your documentation is aligned with EU standards.
It covers all key components, from General Requirements needed across documents to ensuring GSPR Conformity. The checker also supports robust Post-Market Surveillance (PMS) and Post-Market Clinical Follow-Up (PMCF) processes, ensuring your products remain compliant even after they hit the market. Additionally, it assists with Clinical Evaluation and Risk Management, two crucial elements of your compliance strategy. Our checker is also equipped to handle Other Regulatory Processes and the preparation of Annex II Technical Documents, ensuring no detail is overlooked. Plus, it can help navigate Special Cases, making it the ultimate tool for comprehensive EU MDR compliance. With all these components integrated into one solution, you can be confident in your ability to meet the ever-evolving regulatory standards.
With our free EU MDR Compliance Checker, you can simplify the complexities of regulatory compliance and ensure your products meet the highest standards. Stay ahead of the curve and safeguard your business by making compliance effortless and efficient every step of the way. Download it free today.
Flat Monthly Fee Regulatory Consulting
With Mantra Systems’ flexible regulatory consulting options, we can tailor pricing to suit your MDR compliance needs. As well as traditional Fixed Fee Consulting, we also offer a Fractional Regulatory Consulting option for companies that would prefer lower initial costs and no long-term commitment.
Book a free consultation to discuss this alongside your regulatory needs with our team.












































































































