

A Mantra Systems x Cyber Alchemy Perspective - Episode 3
This article is published in partnership with Cyber Alchemy. Mantra Systems specialises in medical device regulatory strategy and technical documentation for UK and EU MDR/IVDR pathways. Cyber Alchemy focuses on cybersecurity, helping teams develop and evidence security for software-enabled and connected medical devices. Together, we're producing a practical series for MedTech teams: what to build, what to defer, and how to avoid rework when moving between UK, NHS procurement, and EU routes.
Reviewers do not assess your security. They assess your evidence of it.
The pattern we see most often in late-stage submissions: the security is fine, sometimes seriously impressive, but the technical file presents it as a mix of tool screenshots, generic policies, a penetration test PDF stapled in near the back, and a sentence that says, in effect, “we take security seriously.” The reviewer cannot work out, in a reasonable amount of time, what threats the product faces, what controls address them, and what evidence proves those controls work. So they ask. And ask. And the clock keeps moving.
This is what this article is about. Episode 1 covered what minimum viable cybersecurity evidence looks like across UK, EU and NHS routes. Episode 2 covered why nonconformities happen and how to recover. This article sits between the two: it is about the controls themselves, and specifically about how to present them so they read as evidence, quickly, traceably, and without inviting the kind of questions that turn a six-week conformity assessment into a six-month conversation. It is also the cybersecurity companion to our joint Webinar #1 with Mantra Systems – From Nonconformity to Approval.
Key takeaways
- Good controls in a bad evidence format generate findings. Reviewers cannot give you credit for security work they cannot follow. The fastest improvement in most files is structural, not technical.
- Three properties make controls read well: scoped (the boundary and assumptions are explicit), traceable (threats map to controls map to verification), and navigable (a reviewer can find anything in minutes, not days). This article explains how to evidence each of them.
- Five artefacts do most of the work: a boundary and interface inventory, a threat-to-control-to-verification table, an SBOM coverage statement linked to vulnerability triage, a pen-test scope rationale tied to the device boundary, and an access control and audit logging narrative. Get those five right and the rest is easier.
- The most common findings are documentation gaps, not security gaps. Reviewers raise issues because they cannot follow the evidence, not because a control is missing, so the fix is usually to re-shape work you have already done.
- The artefacts that get controls past a reviewer are the same ones that survive post-market, which is the subject of Episode 5 of this blog series. Build them once, in a maintainable shape, and they keep paying back.
Companion perspective (Mantra Systems)
This article focuses on the cybersecurity side: how to evidence security risk controls so a notified body, the FDA, or an NHS assessor can follow the logic from threat to verification quickly. The companion piece from Mantra Systems covers how the same principle applies across design controls, clinical risk controls, usability controls, and post-market surveillance: the wider risk control story your file needs to tell.
- Read Mantra Systems’ expert perspective: Understanding Risk Management for SaMD
- Book a joint Cyber Alchemy × Mantra Systems review: Book now
Why this matters now
1) Security evidence is being asked for earlier in the lifecycle
Five years ago, a software-enabled medical device could often reach pilot with a high-level security policy, a one-off penetration test, and a verbal assurance that “the cloud is secure because AWS is secure.” That window has closed. Procurement teams now send detailed security questionnaires before clinical sign-off. NHS trusts run DTAC due diligence at pilot stage. Investors commission third-party diligence at Series A. And under EU MDR Annex I (GSPR 17.2 and 17.4), notified bodies increasingly look for explicit minimum IT security requirements for the intended environment, plus a state-of-the-art lifecycle that includes information security. The same artefacts now need to satisfy more audiences, more often, and earlier, and be presented well enough that none of them asks the same question twice.
2) “What does good look like?” is a regulator question now, not a vendor one
IEC 81001-5-1 (security activities in the product lifecycle), AAMI TIR57 (risk management for security) and MDCG 2019-16 (medical device cybersecurity guidance) all converge on the same expectation: that security risk controls are the visible output of a defined lifecycle process, traceable to threats, verified, and maintained. The FDA’s Section 524B premarket guidance asks for substantially the same evidence story in different vocabulary. DTAC v2.0 (from 6 April 2026) sharpens what NHS procurement expects to see. The frameworks differ in detail but agree on the spine: threats, controls, verification, residual risk, post-market.
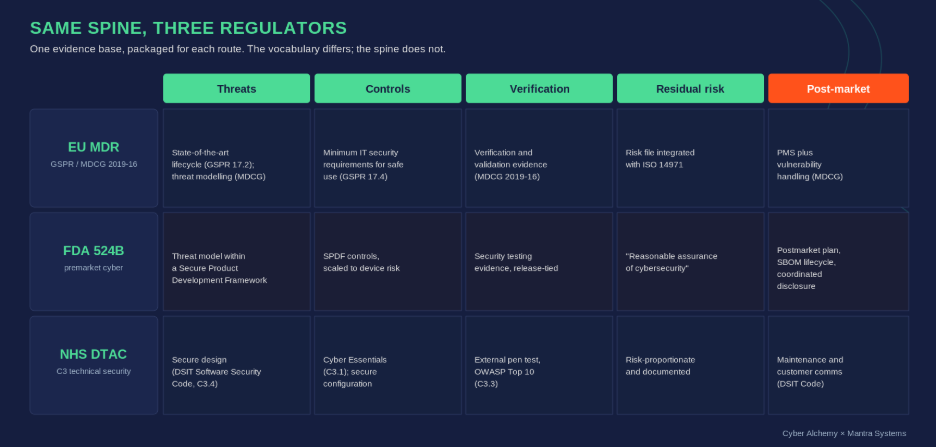
The same evidence base, mapped to EU MDR, FDA 524B and NHS DTAC. Build it once, then package for each route.
For a fuller side-by-side of what each market expects, and where the evidence overlaps, see Cyber Alchemy’s Access to Markets guide.
3) This article sits inside a recovery arc
In the previous article we wrote that the gap between security that exists in the product and security that exists in the documentation is almost always closable, but it takes structured, methodical work. This article is the structural template for that work: what reviewer-readable looks like before there is a finding to recover from, and the shape a corrective action plan tends to take if there is one.
What good looks like
Security risk controls read well when three properties are true at once.
Scoped. The boundary of the device, what is in scope, what is out of scope, and what assumptions you are making about the environment, is explicit on the first page a reviewer reads, not implicit in an architecture diagram on page 47. Most disputes about controls are actually disputes about boundary. If a reviewer thinks the device includes the hospital network and you do not, your controls will look incomplete to them and excessive to you. Resolve this in writing, up front.
Traceable. A reviewer can follow a single line from a credible threat, through the control that addresses it, to the verification evidence that proves the control works, to the residual risk that remains. The line is short, the references are stable, and the artefacts referenced actually exist where the document says they do. Traceability is the spine of “state of the art” under MDCG 2019-16 and the spine of “reasonable assurance of cybersecurity” under FDA 524B. It is the same idea.
Navigable. A reviewer can answer a specific question (how is patient data protected in transit? what happens if an update fails? who can change device configuration?) in minutes, by going to a named artefact. The index works. The references resolve. The pen-test scope matches the boundary diagram. The SBOM matches the build. The logging description matches what the system actually emits.
A useful litmus test: hand the file to a smart engineer outside your team and ask them to summarise your security posture in a paragraph and point to the evidence behind each claim. If they can produce something reasonable in thirty minutes, the file is well-shaped. If they cannot, you probably have a presentation problem rather than a controls problem, which is good news, because presentation problems are faster to fix.
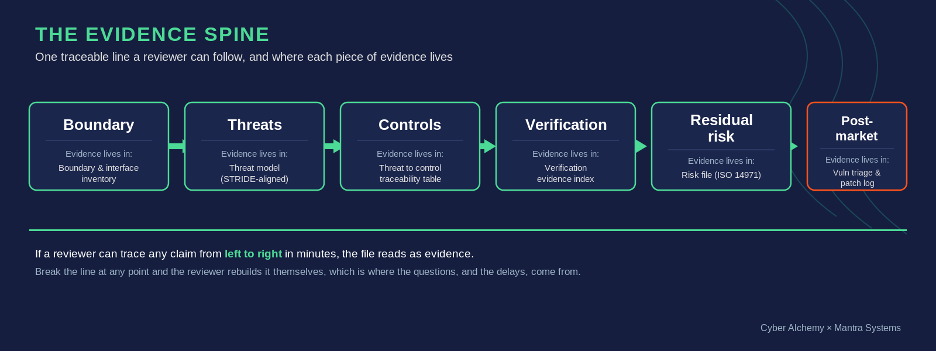
The evidence spine: a single traceable line, and where each piece of evidence lives.
The minimum artefacts list
For most software-enabled and connected medical devices, the minimum set of artefacts that makes security controls read well is short. Each answers a question a reviewer is going to ask. The point is to have one named, versioned, releasable artefact per question, not the same content scattered through ten documents.
- Boundary and interface inventory. Names every external interface, every trust boundary, and every shared-responsibility line.
- Threat model summary focused on credible abuse cases for the intended environment. STRIDE-aligned is great. What matters is that the threats are real for your device.
- Threat to control to verification traceability table. The single most useful document in the file. Top threats, with stable IDs, one sentence per column.
- Verification evidence index. Every piece of test evidence referenced by a stable ID, tied to a product version, with an owner.
- Penetration test evidence summary. One page in front of the report: scope rationale, what was in and out, remediation, retest status, and limitations. An overview of the company that did the assessment and their qualifications e.g. CREST accreditation, is also useful to demonstrate quality.
- SBOM coverage statement plus a vulnerability triage log with one worked example. The SBOM is not the evidence; the worked example showing detection → assessment → decision → closure is.
- Access control and audit logging narrative. Roles, privileged actions and how they are protected, what is logged, who can read the logs, retention, tamper protection.
- Secure update and patch policy statement that is honest about what you can deliver.
That is the spine. If those eight artefacts are present, versioned, internally consistent, and cross-referenced, most of the questions a reviewer will ask have answers waiting for them.
Common failure patterns
These are the patterns we see most often when teams ask us to look at a file before submission, or after a finding has arrived. None of them are about bad security. They are about evidence formatting.
Controls are described as intentions, not testable behaviours
“We use encryption.” “Access is restricted.” “Logs are protected.” A reviewer cannot do anything with these sentences. They are statements of intent, not specifications of a control. A control should be written as something testable: what is encrypted, with what algorithm, with what key management, verified by which test, with the evidence stored where. Three short clauses turn an intention into a control.

Three short clauses, what it is, how it works, and how it is verified, turn an intention into a control.
Penetration test scope does not match the device boundary
The most expensive mistake we routinely find. A test was commissioned in good faith, often by a capable testing firm, but the statement of work scoped it to the cloud APIs and ignored the mobile app, or scoped it to the mobile app and ignored the firmware update path, or scoped it to the development environment and not the production build. The report is real, but it does not test the boundary in the technical file. Pen tests should follow boundary alignment, not precede it; the scope must read like a paragraph from your technical file.
SBOM exists but does not connect to anything
When Log4j was disclosed in late 2021, manufacturers with an automated, release-tied SBOM answered the “are we affected?” question in minutes. Manufacturers with an SBOM exported to a PDF eighteen months earlier answered it in weeks. An SBOM by itself is not evidence; the evidence is the chain:
coverage statement → CVE monitoring workflow → triage log → at least one worked example from detection to closure.
Under both EU MDR and FDA 524B the SBOM obligation runs through the product lifecycle, not just to the point of submission.
Logging is implemented but not described
A surprising number of files implement excellent audit logging (privileged actions logged, retention defined, tampering protections in place) and then describe none of it. From a regulatory evidence perspective, an undescribed log is the same as no log. The fix is one page: what events are logged, why those events, who can read the logs, retention, and how the logs are protected from modification.
Access control is implied in the architecture but not evidenced
“Role-based access control is in place” is a sentence, not a control. The control is the role catalogue (clinician, patient, administrator, support engineer) with allowed actions per role; with privileged actions explicitly named (configuration change, user management, remote support, data export); with how those actions are protected (MFA, step-up auth, time-bound access, approvals); and with where this is verified. Five lines on a page, consistently formatted across every privileged pathway.
“Future-state” controls written as if they were current
The one that turns a manageable conformity assessment into a non-conformity. A control is described in the file in the form it will take after the next release, not as it currently behaves in the shipped product. The reviewer tests against the file. The behaviour does not match. As Episode 2 covered, missing your own stated controls is how most cybersecurity nonconformities happen. Write controls that describe the release being submitted, and use change control to update them when the product changes.
Practical steps: five patterns that pay back fast
The fastest way to improve reviewer readability is to take a small number of high-leverage controls and present them in a consistent evidence format. The patterns below are the ones that usually deliver the biggest reduction in questions. None of them require new security work. They re-shape work that is, most of the time, already done.
Pattern 1: threat to control to verification traceability
The single most useful document in a well-shaped technical file is a traceability table that connects each material threat to the control that addresses it and the verification evidence that proves the control works. Done well, it removes most of the follow-up questions reviewers ask, because most of those questions are reviewers reconstructing this table for themselves. Every row has the same shape:
- Threat ID and one-sentence description (“T-014: tampering with patient identifiers in transit between mobile app and cloud API”).
- Risk rating, using the same scale as the wider device risk file so it lines up with the clinical risk language.
- Control ID and one-sentence description (“C-022: mutual TLS with certificate pinning and short-lived tokens between mobile app and API”).
- Verification reference (“V-022.1: pen test 2026-03, finding 4 retested 2026-04, see Annex C”).
- Residual risk note: what is left, why it is acceptable, and what monitoring catches drift.
Three rules make the table actually useful. IDs are stable across releases, so a reviewer comparing this submission to the previous one can see what changed. Verification references resolve, so clicking V-022.1 takes you to the actual test record. And “verified by inspection” is fine as an evidence type, but it has to point at a specific inspection record with a date, an inspector, and a result. Threat modelling itself should align with a recognised framework (STRIDE is the most common and pairs well with AAMI TIR57), but the framework matters less than the discipline of producing threats that are credible for your intended environment.
| Threat | Risk | Control | Verification | Residual risk |
|---|---|---|---|---|
| T-014: Tamper with patient IDs in transit (app to cloud API) | High | C-022: Mutual TLS, cert pinning, short-lived tokens | V-022.1: Pen test 2026-03, finding 4 retested 2026-04 (Annex C) | Low; TLS-error alerting |
| T-031: Unauthorised config change by support engineer | High | C-040: Step-up MFA + time-bound, approved config access | V-040.2: Access review 2026-02; config-change log (Annex D) | Low; quarterly recert |
| T-008: Known CVE in third-party library reaches production | Med | C-015: Release-tied SBOM + CVE triage workflow | V-015.3: Triage log CVE-2026-0412, closed 2026-04 | Low; continuous CVE monitoring |
| T-022: Failed update leaves device in unsafe state | High | C-051: Signed updates, auto rollback to last-good image | V-051.1: Release test on representative hardware, build 4.2.1 | Low; rollback verified each release |
| T-045: Audit logs altered to hide misuse | Med | C-033: Append-only, access-controlled logs with integrity checks | V-033.2: Config snapshot + tamper-alert test (Annex E) | Low; integrity alerts monitored |
Example threat-to-control-to-verification table. Stable IDs, one sentence per column, references that resolve.
Pattern 2: SBOM and vulnerability management as a workflow, not a document
The SBOM is not the evidence the reviewer wants to see. The workflow around the SBOM is. Three things, in this order: a coverage statement (what is in, what is excluded and why, how it is generated, build-time, release-tied, in SPDX or CycloneDX, and how often it is regenerated); a CVE monitoring workflow (which feeds, who triages, against what criteria, such as exploitability in your deployment context, safety impact, realistic exposure, and availability of mitigation short of a code change); and a triage log with at least one worked example from detection to closure.
The worked example carries more evidential weight than the policy. A reviewer who reads “CVE-2025-XXXX was assessed on date X; rated low-exploitability for our deployment because Y; mitigated by Z action; verified by retest on date W; closed by [name]” learns more about your vulnerability management than any number of policy paragraphs. A maintained SBOM connected to a CVE monitoring workflow is also the foundation of how you survive the next Log4j-class event, which Episode 5 will explore in the post-market context.
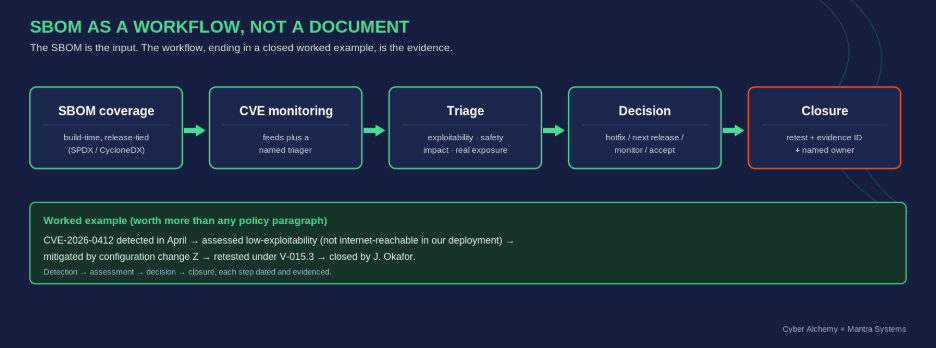
From SBOM coverage to a closed, dated worked example. The workflow, not the document, is the evidence.
Pattern 3: penetration test evidence formatting
A pen test report on its own is almost never a piece of evidence in the form a reviewer wants. The report is the raw material. The evidence is a one-page summary in front of the report that does four things:
- States the scope and the rationale for it. “The test covered the cloud APIs, mobile application and OAuth flow between them, against build 4.2.1. It did not cover the firmware update path, which is covered by a separate vendor-supplied attestation under reference V-031.”
- Summarises findings by severity, with one sentence per high or critical: what it was, what was done, when it was retested, with the retest evidence ID.
- States the limitations of the test honestly. Time-boxed engagements have limits; black-box has limits; grey-box has different limits. Saying so prevents the reviewer concluding the limits themselves and asking about them.
- Connects retest evidence to the original findings. The retest is what closes the finding, not the original report.
Two anti-patterns to avoid. Do not embed a 60-page report in the technical file body; reference it as an annex with a stable ID and put the summary at the front. And do not paste raw tool output (CVSS scores from a scanner, screenshots from a fuzzer) as evidence on its own; tool output is data, not interpretation. On cadence, there is no universal interval; “annual plus triggers” (major release, new integration, significant architectural change, new class of vulnerability) is the framing that holds up under most reviews. Document the cadence and stick to it.
Pattern 4: access control and audit logging that read well
Two of the most asked-about areas in any security review, and both easier to evidence than they look. Start with a role catalogue. Name every role the system supports and the allowed actions per role; be specific. “Administrator” is not a role; “tenant administrator who can create users and reset MFA but cannot read patient data” is a role. If a single human can become any role at any time, that is a privilege escalation path and it needs to be named.
Then list privileged actions explicitly (configuration change, user management, remote support session, data export, key rotation, code deployment to production), and for each, how the action is protected (MFA, step-up auth, approval workflows, time-bound access, break-glass procedures) and how it is logged.
The audit logging description is one page: what events are logged, why, where logs are stored, who can read them, retention, how they are protected against modification, and how tampering would be detected. The same page works in front of DTAC, EU MDR, FDA, and procurement diligence. Evidence the design with two artefacts: the one-page narrative, and a verification record (a configuration snapshot from the release under review, plus a sample of log output demonstrating the events described actually appear).
| Field | Worked example: privileged action “remote support session” |
|---|---|
| Claim | Remote support sessions are restricted, authenticated, time-bound and fully logged. |
| Control design | Engineers request access via an approval workflow; access is role-scoped (no patient-data read), protected by step-up MFA, time-boxed to 60 minutes, and auto-revoked. Each session opens an audit record. |
| Verification | Configuration snapshot from build 4.2.1 showing session limits; sample session log demonstrating start, actions taken, and revocation. |
| Evidence IDs | C-040 (control), V-040.2 (verification record), Annex D (log sample). |
| Limitations / assumptions | Assumes the customer manages their own identity provider and endpoint security. Break-glass access is separately logged and reviewed within 24 hours. |
Access control evidence template, filled in for one privileged action. Use the same shape for every privileged pathway.
Pattern 5: secure updates and patching, evidenced without overpromising
This is where files most often write a cheque the post-market team cannot cash. Episode 2 covered why ambitious patch policies turn into nonconformities; the controls side of the same problem is to write update and patch evidence that reflects what your operating model actually delivers. Four short artefacts cover most of what reviewers want to see:
- A one-page update mechanism summary: what is signed, how signatures are verified, how keys are managed, and how the device behaves during and after an update.
- A rollback or safe-state statement: if an update fails, what does the device do, and how is the user informed. Honest answers are far more credible than aspirational ones.
- Patch decision criteria: severity tier, exposure in deployment context, safety impact, mapped to an action path (hotfix, next release, monitor with mitigation, accept). The criteria link directly to the vulnerability triage log.
- Verification references: CI/CD checks demonstrating signing in the build, release test evidence demonstrating a sample update completing on representative hardware, and one worked example of an update verification record.
On timelines: EU MDR (via MDCG 2019-16) says “timely”. FDA Section 524B says “reasonably justified regular cycle” for known unacceptable vulnerabilities and “as soon as possible out of cycle” for critical vulnerabilities causing uncontrolled risk. DTAC frames expectations through Cyber Essentials and the DSIT Software Security Code of Practice. None of these prescribe a number. Whatever you state in your own QMS or technical documentation becomes the number you are audited against. Pick something you can sustain in clinical deployment.
| Severity | Exposure in deployment | Safety impact | Action path | Evidence produced |
|---|---|---|---|---|
| Critical | Exploitable in clinical deployment | Could affect patient safety | Out-of-cycle hotfix, as soon as possible | Triage entry + emergency release record + retest |
| High | Reachable, mitigations available | Limited or indirect | Expedited or next scheduled release, per triage | Triage entry + release test evidence |
| Medium | Not exposed in current configuration | None identified | Monitor with documented mitigation; fix in routine release | Triage entry + mitigation note |
| Low | No realistic exposure | None | Accept with rationale; review on change | Triage entry + residual-risk rationale |
Patch decision criteria mapped to action paths and the evidence each path produces.
Related: Webinar #1, From Nonconformity to Approval
The practical patterns in this article are the same ones we walked through in Webinar #1, our joint session with Mantra Systems on recovering from notified-body nonconformities and preventing them from recurring. The webinar covered the integrated clinical-and-cyber recovery blueprint, including a worked turnaround story, the seventy-two-hour triage steps, and the artefacts that turn a finding into a closed file.
Watch the recording on LinkedIn or YouTube.
Free downloads
- System Boundaries: Who Controls What (PDF)
- A one-page reference for one of the most common evidence problems in connected-device files: where your security responsibility ends and the customer's begins. Covers device and firmware, mobile applications, cloud and backend, integrations (EHR and APIs), and shared responsibilities, with the explicit assumptions that need to be written into your technical documentation. The example boundary diagram on the second page is the shape we recommend most teams adopt.
- 10 Core Procurement Artefacts (PDF)
- The checklist we use to keep procurement and assurance work from becoming a last-minute scramble. It covers the ten artefacts most frequently requested across DTAC, EU MDR and private procurement, including security architecture and boundaries, threat model and risk register, the threat-to-control-to-verification traceability matrix discussed in this article, SBOM and vulnerability monitoring, a VDP/PSIRT route, patch policy, security test evidence, logging and monitoring approach, supplier security controls, and incident response, plus a suggested cadence for keeping them current.
Companion perspective and next step
Mantra Systems’ companion article walks the same idea across the wider risk control story (design controls, clinical risk controls, usability controls and post-market surveillance), and shows how reviewers expect those controls to tie together in the technical file. Read both to get the full picture of controls as evidence rather than controls as paperwork.
Read the companion perspective from Mantra Systems – Understanding Risk Management for SaMD
Next step: book a joint review
If you want an evidence-first view of where your security risk controls currently stand against EU MDR, FDA Section 524B and NHS DTAC, and a clear sense of what to fix first, book a joint review with Cyber Alchemy and Mantra Systems. In your free 30 minutes review, we will:
- Clarify your system boundary, intended environment, and the assumptions your technical documentation currently implies.
- Identify the top three cybersecurity assurance gaps in your current evidence, and how they connect to the wider clinical and regulatory story.
- Agree the minimum artefacts and verification evidence needed for the route you are targeting, so you do not overbuild for markets you may never enter.
- Turn the above into a short, prioritised action plan you can execute over the next two to four weeks.
FAQs
-
It means presenting your security risk controls as a traceable chain a notified body can follow without prompting: from a credible threat (in line with MDCG 2019-16 and GSPR 17.2/17.4), to the control that addresses it, to the verification evidence that proves the control works, to the residual risk that remains. The controls themselves are not new; what makes them evidence is that the chain is explicit, stable, and tied to the release on the market.
-
Substantially yes, in different vocabulary. FDA 524B asks for a Secure Product Development Framework, a release-tied SBOM with lifecycle vulnerability monitoring, and a defined approach to vulnerability disclosure and patching. EU MDR (through GSPR 17.2/17.4 and MDCG 2019-16) asks for a state-of-the-art lifecycle, threat-to-control traceability, and demonstrable verification. The spine is the same: if your evidence is shaped right for one, it is mostly shaped right for the other, with packaging differences.
-
DTAC (technical security under the C3 domain, with the v2.0 form required from 6 April 2026) leans on Cyber Essentials and the DSIT Software Security Code of Practice. The artefacts that satisfy a notified body or the FDA (boundary, traceability table, SBOM and vulnerability triage log, pen-test summary, logging and access control narrative) also satisfy most of what an NHS assessor needs, with a DTAC-specific summary in front. Build the truth once; package it for the audience.
-
STRIDE is a sensible starting point and one we always recommend due to its versatility and ease of use. PASTA, LINDDUN (for privacy-heavy systems), or attack trees can be appropriate depending on the device. The framework matters less than the discipline of producing threats that are credible for your intended environment, with a stable ID per threat, and a clear path into your control set.
-
There is no universal interval. Annual is a reasonable floor; 'annual plus triggers' (major release, significant architectural change, new integration, new class of vulnerability) is the framing that holds up under most reviews. A defensible cadence you actually meet is worth more than an aspirational one you miss.
-
No. The artefacts described here are the same ones you would build during a corrective action plan, and a pre-emptive evidence pass before review questions arrive is usually faster than reacting to them. Most teams find that one to two weeks of structured work shifts a file from answering questions to anticipating them, which is the difference between a long review and a short one.
About the author
Luke Hill, Senior Security Consultant at Cyber Alchemy
Luke brings deep expertise in security consultancy, penetration testing and regulatory‑aligned security measures in the Health and Social Care sector. He leads Cyber Alchemy’s technical and regulatory efforts in the MedTech space, supporting a broad range of MedTech companies in building resilient devices and applications that comply with complex UK, US, and EU regulations.
Related
- Case study: See how Cyber Alchemy supported Adaptix
- Essential Guide to SaMD and MedTech Device Security: a plain-English overview of how cybersecurity fits across the SaMD and connected-device lifecycle.
Articles in this series
- Episode 1 from Mantra Systems
- Where to Launch First? A MedTech Founder's Regulatory Roadmap to the EU, UK and US
- Episode 1 from Cyber Alchemy
- EU MDR & NHS DTAC Cybersecurity Requirements for UK Market Entry
- Episode 2 from Mantra Systems
- How to handle non-conformities and get back on track
- Episode 2 from Cyber Alchemy
- EU MDR, FDA 510(k) and DTAC Cybersecurity Nonconformities: How to Recover
- Episode 3 from Mantra Systems
- Understanding Risk Management for SaMD
- Episode 3 from Cyber Alchemy
- Security Risk Controls in the Medical Device Technical File
















































































































{kind=link}