

All medical devices have inherent risk. Under the EU Medical Device Regulation (EU MDR 2017/745), risk should be assessed in line with ISO 14971.
Risk is defined as:
…the combination of the probability of occurrence of harm and the severity of that harm.
In medical device risk management, this involves assessing the likelihood of sequence of events leading to a hazardous situation and the probability that the situation results in harm, alongside the potential impact on the patient.
These factors are evaluated against predefined risk acceptability criteria, often using a structured approach such as a risk matrix, to determine whether an individual risk is acceptable or if additional control measures are required. This systematic process supports consistent decision-making, ensures transparency, and maintains traceability throughout the lifecycle of risk management activities.
But identifying clinical and design risks is only part of the picture. What happens when those risks intersect with cybersecurity threats — and how do you evidence controls that satisfy Notified Bodies?
A Mantra Systems x Cyber Alchemy Perspective - Episode 3
This article is published in partnership with Cyber Alchemy. Mantra Systems takes medical devices through UK and EU MDR/ IVDR, from regulatory strategy to technical documentation. Cyber Alchemy focuses on cybersecurity, helping teams develop and evidence security for software-enabled and connected medical devices. Together, we're producing a practical series for MedTech teams: what to build, what to defer, and how to avoid rework when moving between UK, NHS procurement, and EU routes.
Software Risks: Where Should You Start?
As well as looking at individual risks in isolation, it is important to consider the overall residual risk of the device. This will inform the overall risk of the device when used as intended (or misused) within its clinical setting.
A methodical approach to risk assessment is crucial to ensure that there are no unresolved risks, ensuring accurate and comprehensive assessment of the overall risk of the device. This is especially important for software as a medical device (SaMD), which is usually formed of multiple functional units and storage components and may also incorporate artificial intelligence (AI) features.
Risks can arise from any part of the device’s function. From user interaction to the device’s output, there is a possibility of certain risks arising from individual components. When these components are combined to form the device, the number of risks can increase significantly.
It is therefore important to consider the function of each unit/component of the device, what could go wrong with this feature, and how this fault would impact the function of the device.
Example
An ambient AI scribe may use a cloud storage system for storing patient data. If the link between the cloud service and the device fails, then data might not be stored correctly, risking loss of patient data. The loss of patient data could then result in incomplete medical records which are used to make clinical decisions.
You can then determine the potential harms that could occur to the patient as a result of this. Thus, it's crucial to assess all components of the device, how they impact the function of the device, and ultimately what can happen if this component fails.
Adopting a structured approach is vital to ensure all possible risks are identified. This can be approached by creating a device traceability workflow. Starting with input to the device, map out the individual units/components that comprise the device, and how these link together to produce the output of the device. Producing a structured device workflow can provide insight into where risks can arise.
Mapping out the flow of the device's units and components helps identify potential issues that can occur within the device's workflow and how that would impact the intended use of the device and subsequently cause patient harm.
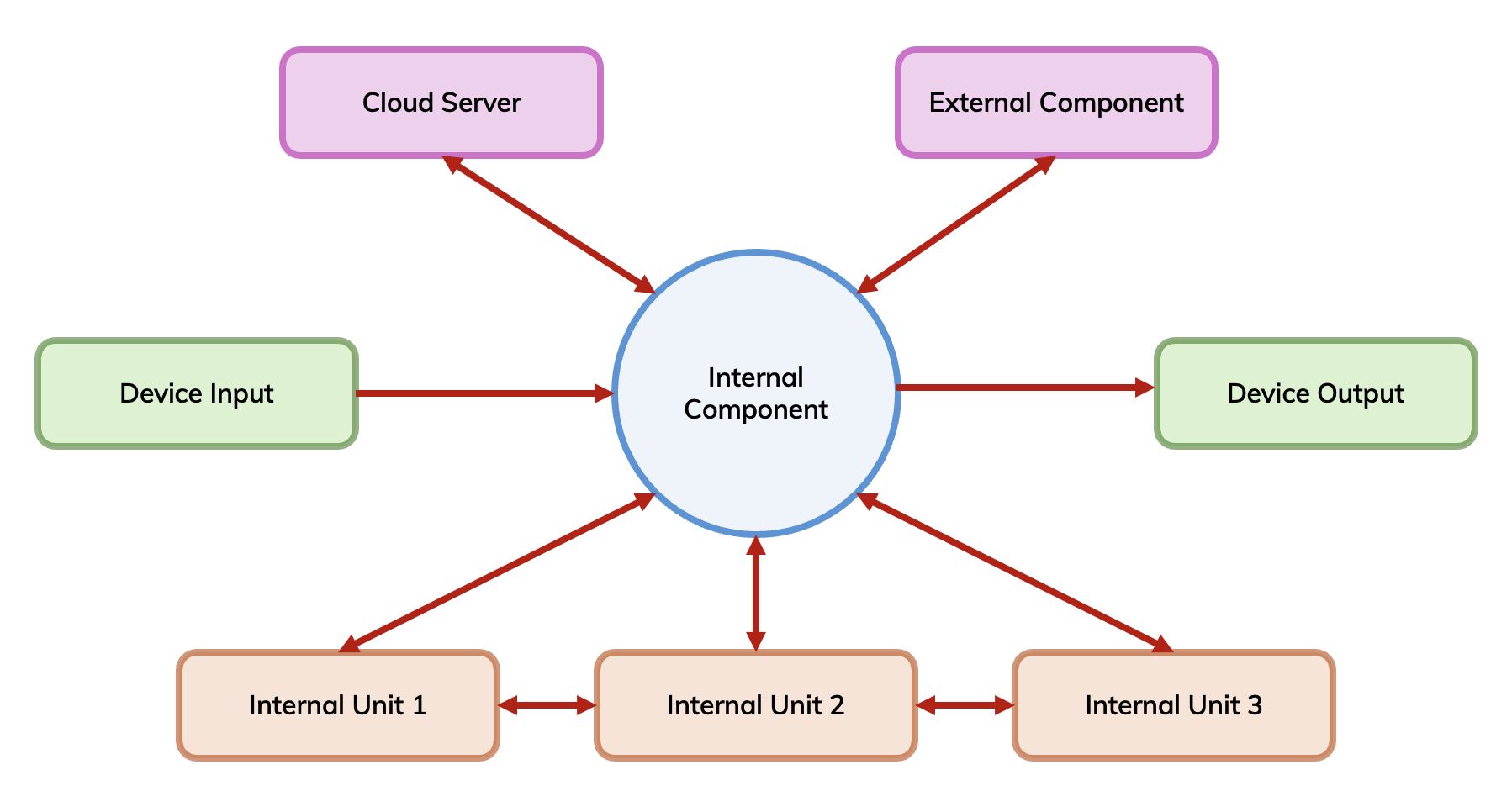
Risks tend to fall into categories, such as:
- User-related issues
- What would happen if the user were unable to use the device?
- User-related misuse
- What would happen if the user used the device incorrectly?
- What would happen if the device were used in the incorrect patient population or clinical field?
- Software design risks
- How is the device input processed to produce the device output?
- What would happen if there was an issue with the device input?
- What if a device unit/component fails?
- Data storage risks
- How is data stored by the device?
- What would happen if this unit failed?
- What would happen if Internet access were lost?
- Cybersecurity and access risks
- What would happen if an unauthorised user gained access to the device or its data?
- What vulnerabilities could be exploited through the device’s network connections or third-party integrations?
- How would a security breach impact the integrity of the device’s output or patient safety?
To mitigate against these risks, the device manufacturer must implement risk control measures that are appropriately designed to mitigate the particular risk.
Clinical Risk Controls
Risks that arise from clinical use (or misuse) can be approached using clinical-based risk controls. Here’s an example of a clinical risk and how it can be mitigated using a clinical risk control.
Example
Imagine an SaMD device used to assess the probability of surgical intervention in gastroenterology. The device should only be used in gastroenterology but this may not be immediately clear to clinicians, which may result in the device being used in a different clinical field to assess surgical intervention.
This is where robust and well-structured instructions for use (IFU) is valuable. An IFU provided to the user - in this case the clinician - should display the:
- key use cases of the device
- device’s intended purpose
- patient population the device is intended for
- contraindications of the device
The contraindications would state when the device should not be used to ensure the device is used within its intended clinical field and patient population and by the correct intended user.
Device Usability: What can go wrong?
Risk controls can also incorporate the usability of SaMD. This refers to how the software is designed to intrinsically mitigate risk.
Including design components that reduce the likelihood of a hazardous situation occurring is essential to ensure your device is safe and works as intended.
Example
A common example for software devices is access security. SaMD often stores patient data to perform its intended purpose. Therefore, there is an inherent risk of security issues associated with unauthorised access to patient data. If patient data security is compromised, this can result in several different harms, such as loss of data or unauthorised sharing of patient information.
This is where device usability can be assessed to mitigate this risk. For example, incorporating more robust security measures, such as two-factor authentication, to reduce the likelihood of unauthorised access to patient data. Therefore, it is important to consider intrinsic design features of a device to ensure risks are mitigated.
Ensuring Traceability of Risk Controls
Beyond identifying and implementing risk control measures, it is essential to ensure clear traceability of these controls within the technical documentation.
Traceability demonstrates how each identified risk is linked to specific control measures, and how those controls are verified and validated to be effective. Within the technical file, this typically means maintaining structured links between the following:
- Risk Management File
- Design Documentation
- Verification & Validation Reports
- Usability Engineering Records
- and Post-Market Surveillance plans.
This level of transparency allows reviewers to quickly confirm that risks have not only been identified but systematically mitigated and supported by evidence. Without strong traceability, risk controls can appear fragmented or incomplete, even when they are otherwise well designed, leading to additional questions and delays during Notified Body review.
Assessing Device Risk Post-Market
Performing a risk assessment shouldn’t be a task that’s only conducted for market access. The risk of a device should be assessed once the device enters the market and throughout the device’s lifetime, in line with Post-Market Surveillance (PMS) requirements (Article 83).
This is an opportunity to monitor risks identified through risk assessment throughout the device’s lifetime. Risks should be monitored both pre-market and post-market. PMS monitors real-world use of the device to determine whether identified risks are impacted by use of the device in clinical settings.
Example
For a device which utilises AI, an inherent risk of the device is hallucination. This risk should be monitored on an ongoing basis using PMS activities. PMS can be incorporated as a risk control where the real-world use of a device may more greatly inform the likelihood of a hazardous situation occurring.
At the design stage, initial controls might include output confidence thresholds that suppress low-certainty results, mandatory human-in-the-loop review before any clinical action is taken, or clear UI indicators that flag AI-generated content as requiring clinician verification. These design controls establish the baseline risk level before the device reaches market.
For hallucinations, the manufacturer could monitor complaints associated with the device and assess how often a hallucination is reported by a user. Trend analysis should be performed to determine if hallucination rate changes over time. If significant changes in hallucination rate occur over time, root cause analysis must be performed to determine the cause of the increased hallucination rate. Subsequently, appropriate design controls can be implemented to mitigate the risk of hallucination.
This example highlights how a robust PMS system can be leveraged as a risk control to inform the risk of a device associated with real-world use.
Therefore, implementing appropriate and validated risk controls is pivotal to ensure the overall risk of a SaMD is mitigated and the device safety is consistently monitored throughout the device’s lifetime.
Next step: Get a free joint risk review
Risk management for SaMD doesn’t sit neatly in one box. Regulatory gaps and security gaps often overlap — and reviewers notice when they’re addressed separately. Mantra Systems and Cyber Alchemy are offering a free 30-minute joint review to help you identify where those gaps are, before your Notified Body does.
Articles in this series
- Episode 1 from Mantra Systems
- Where to Launch First? A MedTech Founder's Regulatory Roadmap to the EU, UK and US
- Episode 1 from Cyber Alchemy
- EU MDR & NHS DTAC Cybersecurity Requirements for UK Market Entry
- Episode 2 from Mantra Systems
- How to handle non-conformities and get back on track
- Episode 2 from Cyber Alchemy
- EU MDR, FDA 510(k) and DTAC Cybersecurity Nonconformities: How to Recover
- Episode 3 from Mantra Systems
- Understanding Risk Management for SaMD
- Episode 3 from Cyber Alchemy
- Security Risk Controls in the Medical Device Technical File















































































































